
Beyond density functional theory
Density functional theory (DFT) has been a successful tool in the study of the electronic structure of real materials for several decades. There now exists plenty of program packages based on DFT that can calculate material properties accurately and efficiently. There however exists a broad class of materials, so called strongly correlated materials, where the electron-electron interaction heavily influences the electronic structure. DFT has problems in describing these types of materials correctly, since it only includes electron-electron interactions in an average way. Recently methods like dynamical mean field theory (DMFT), stemming from the many-body approach in condensed matter theory, has been combined with conventional DFT methods to improve our understanding of strongly correlated systems. Our group works on this kind of method development.
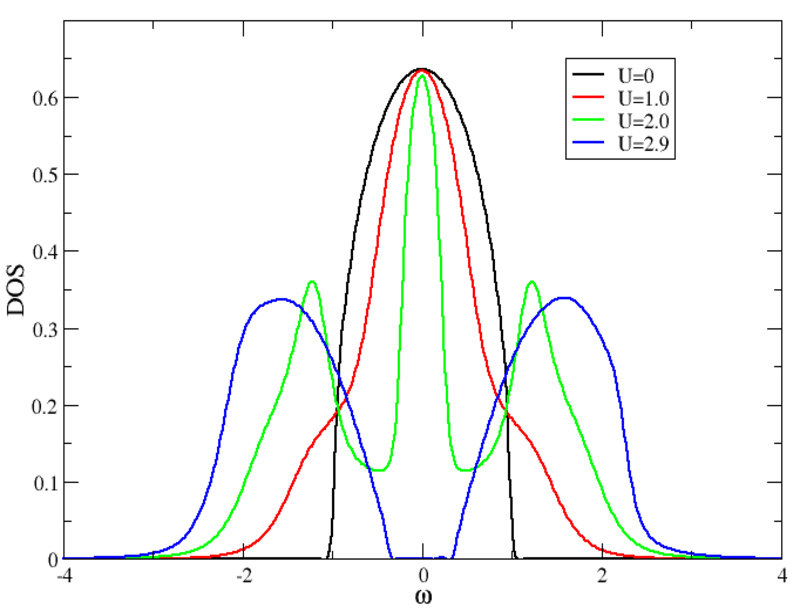
The picture shows the density of states of a model Bethe lattice for different values of the Coulomb repulsion U between electrons, obtained using DMFT. As U is increased, the system goes from a metallic to an insulating state.
Contact
Andreas Östlin